Protomata-Learner version 2.0
Given a sample of (unaligned) sequences belonging to a structural or functional family of proteins, Protomata-Learner infers automata characterizing the family. Automata are graphical models representing a (potentially infinite) set of sequences. They can be used to get new insights into the family, when classical multiple sequence alignments are insufficient, or to search for new family members in the sequence data banks, with the advantage of a finer level of expressivity than classical sequence patterns (such as PSSM, Profile HMM, or Prosite Patterns) enabling to model heterogeneous sequence families.
Quick start guide to Protomata-Learner:
Prepare sequence sample.
To obtain better results, quality is here more important than quantity. Prefer sequences whose membership to the family has been established experimentally and discard suspicious sequences. In order to speed up the program and avoid bias, we advise you also to reduce redundancy in your sequence set by setting to 90% (or eventually less) the maximum allowed redundancy threshold.
Run Protomata-Learner.
Default parameters have been chosen as a good compromise, but if you want a characterization based on "domain similarity", you can raise the maximal size of fragments to 20 or more in order to favour the emergence of longer blocks. On the contrary, if you think that characterization should rather be done on the basis of "amino-acid similarity", you can lower the maximal size of fragments to 10 or less to get shorter blocks.
The significativity of fragment similarity is the main parameter to play with and affects mainly the height of the blocks. The default value of 5 for a maximal size of 15 will consider only significant fragment pairwise alignments. If the signature that you are searching is less conserved, try 3 or 1...
Filling the email field allows you to get an email when the job is finished with an url to retrieve the results. This can be particularly useful if you have to close your browser while the job is still running.
Look at results and adjust quorum if needed.
To view automata and related alignments, several formats are available (opening links in new windows can be convenient for seeing multiple results). Generate new protomata with different quorums to adjust the level of details if you want/need.
If you can not obtain satisfactory results, go back to the first page and try new "Partial Local Multiple Alignments" parameters.
Use automata to search for new sequences in public data banks or a personal set of sequences.
Default pseudocounts are rather conservative and will find closely related sequences. If you want to find more distant sequences, you can generate a new protomaton by selecting, for instance, the dist.20comp set of components in the drop-down list (don't forget eventually to set also the desired quorum). Once it will be generated, follow the corresponding "scan" link.
efore scanning a full data bank, you need to estimate the right threshold score to avoid retrieving too much false hits: scanning a personal set of sequences including sequences from your family of interest but also sequences not from the family but close enough (for instance the first false hits using blast) will give you a clue on the score threshold that separates at best the true from the false hits.
Detailed help
Personal parameters
Job title (optional): a name for your experiment.
Mail (optional): your mail address where you want to receive the notification of the end of the job and a link to access to your results.
Training sequences
Paste your sequences: Paste here a representative sample of protein sequences in multiple sequence FASTA (without ambiguous or special code in the protein sequences, i.e. only the 20 amino acids one letter code) for the family that you want to model by a protomaton. Or select a file containing this set of sequences to upload from your computer.
Maximum percentage of identity allowed between sequences: set here the maximum percentage of identity allowed between sequences for the CD-HIT program [1].
Partial Local Multiple Alignements
The first step of Protomata Learner consists in searching for a set of locally conserved regions which are characteristic of the family, resulting in a partial local multiple alignment (abbreviated to PLMA).
You can either set the parameters of the PLMA search or use an existing PLMA file. The latter option allows to use a PLMA saved in a previous session and to skip this time-consuming part of the program. It can also be used to generate Protomata from PLMA obtained by other means than our algorithm. To use an existing PLMA, click on the link Use PLMA (at the right of Training sequences) and then Upload a PLMA file.
The first approach is to set the parameters used to build the PLMA from the sequences:
Threshold for significance of fragments similarity
The search for the blocks of locally conserved regions is based on significantly similar fragment pairs computed by Dialign 2.2.2 [2]. For each fragment pair, a weight score (denoted w in Dialign [1]) related to the significance of the fragment pair similarity can be computed. Only fragment pairs with a higher weight than the threshold parameter (significant fragment pairs) will be considered to build the PLMA blocks.
Consensus
In strong consensus mode, all the fragments in a block are required to be significantly similar to all the other fragments in the block (clique) whereas, in weak consensus mode, two fragment that are not significantly similar may be in the same block as long as it is possible to connect them by a chain of significant fragment pairs (connected component). Strong mode allows usually to get finer results than weak mode but is slightly more time consuming. We advise to begin with weak mode which is usually sufficient and to switch to strong mode if results are not satisfactory. Weak mode should still be better suited for sets of sequences showing evolutionary drift.
Maximal fragment size
Weight function tends to favour longer fragments. By limiting the size of the significant fragments to consider, one can influence the shape of the resulting automata: bigger fragments induce longer blocks and vice versa (note: limiting the maximal size of the fragment does not limit the maximal size of the resulting blocks). Choose bigger values to get characterizations based on domain similarity or else smaller values to get characterizations based on amino-acid similarity.
Some indicative set of parameters
| Significance threshold | Consensus | Max size | |
|---|---|---|---|
| Default | 5 | weak | 15 |
| Quality | 1 or 3 | strong | 15 |
| Domain | 5 or 10 | weak | 20 or more |
| Domain strong | 5 | strong | 20 or more |
| Amino-acid | 3 | weak | 10 |
| Amino-acid strong | 1 or 3 | strong | 10 |
Protomaton
Quorum:
The quorum is the minimal sequences weight in a block required to keep it.
It can be expressed as a percentage of the total sequences weight, or directly as the minimal sequences weight.
A protomaton will be generated for each value that put, separated by spaces or commas.
By default, three automata are displayed with quorums respectively fixed to 3/3, 2/3, 1/3 of the total weight of the sequences
Weight sequences:
If selected each sequence have a weight representing how unique it is in the set. Otherwise, each sequence have a fixed weight of 1.
Pseudocounts:
Set here the components to use for adding a Dirichlet mixture of pseudocounts to the protomatas
Legend for Protomata and PLMA views
Protomata are automata with 3 types of states: characteristic, gap or exception. As for automata, a sequence is accepted (or recognized) by a protomaton if it can be read in its entire length, beginning in the start state and finishing in the end state.
Conventions used to represent the 3 types of states in protomata:
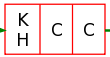
Characteristic region reading a K or a H followed by two C (derived from a plma block with greater support than the quorum).
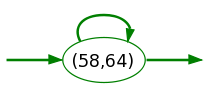
Gap state reading any sequence of amino acid (corresponding to the sequence segments linking the source to the target characteristic states, these segment lengths ranging from 58 to 64 amino acids).

Set of exception paths allowing to read one of the sequence segments linking the source to the target characteristic states, the segment lengths ranging from 73 to 78 amino acids.
PLMA show the alignment of the training sequences by a protomaton.
Each sequence is given a number used to label the transitions related to the sequence. Since, in this view, there is only one sequence per gap or exception state, positions in the sequence are given instead of length of segments:

Positions 1 to 64 of sequence 1 in gap region

Positions 1 to 73 of sequence 4 in exception region
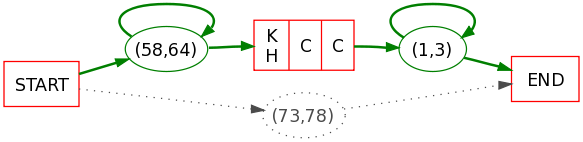
A toy example of Protomaton and associated PLMA:
Protomaton:
PLMA:
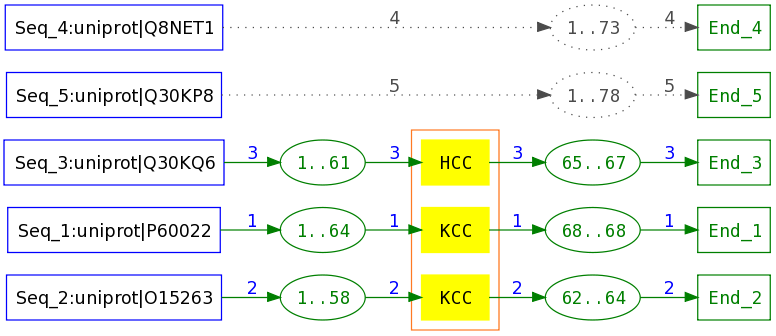
Sequences 1,2 and 3 are accepted through the path constituted of a gap, a characteristic region and a second gap whereas sequences 4 and 5 are accepted as exceptions.
[1] "Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences", Weizhong Li & Adam Godzik Bioinformatics, (2006) 22:1658-9
[2] "DIALIGN 2: improvement of the segment-to-segment approach to multiple sequence alignment" B. Morgenstern, Bioinformatics (1999) 15, 211 - 218.
References
A paper describing Protomata Learner is being written... Protomata Learner improves upon its predecessors Protomata-L and Protomata-CL by a new algorithmic approach but relies on the same fragment merging ideas which were introduced in the following papers:
- Learning Automata on Protein Sequences, François Coste and Goulven Kerbellec, JOBIM 2006.
- A Similar Fragments Merging Approach to Learn Automata on Proteins, François Coste and Goulven Kerbellec, ECML 2005.
In french:
- Problème d'optimisation de recherche de cliques pour caractériser des familles de protéines, François Coste and Goulven Kerbellec, ROADEF 2007
- Apprentissage d'automates par fusions de paires de fragments significativement similaires et premières expérimentations sur les protéines MIP, François Coste, Goulven Kerbellec, Boris Idmont, Daniel Fredouille and Christian Delamarche, JOBIM'04
Contributors
- François Coste
- Andres Burgos
- Anthony Bretaudeau
- Laetitia Guillot
- Thi Hong Hanh Hoang
- Boris Idmont
- Goulven Kerbellec
Thanks also to the authors of Dialign2, GABIOS, ZGRviewer, Graphviz and CD-HIT programs or packages.
Please send questions and comments about Protomata to francois.coste@inria.fr.


